Problem Set 6
Created: 2022-07-16
Tags: #fleeting
Credit
American Express
- 15-digit numbers,
- number starts with 34 or 37
MasterCard
- 16-digit numbers,
- number starts with 51, 52, 53, 54, 55
Visa
- 13 or 16-digit numbers.
- Number starts with 4
Readability
Coleman-Liu Index
S = "average number of sentences per 100 words in the text"
L = "average number of letters per 100 words in the text"
index = 0.0588 * L - 0.296 * S - 15.8
Example
The text the user inputted has
65 letters, 4 sentences, and 14 words.
65 letters per 14 words
is an average of about 464.29 letters per 100 words
because 65 / 14 100 = 464.29
4 sentences per 14 words
is an average of about 28.57 sentences per 100 words
(because 4 / 14 100 = 28.57).
Plugged into the Coleman-Liau formula, a
nd rounded to the nearest integer,
we get an answer of 3
because 0.0588 464.29 - 0.296 28.57 - 15.8 = 3
so this passage is at a third-grade reading level.
-
count the number of letters, words, and sentences in the text
-
Word = any sequence of characters separated by spaces should count as a word,
-
Sentence = period, exclamation point, or question mark indicates end of a sentence
-
Letters = I guess spaces aren't included in letters
average_letters = (letters / words) 100
average_sentences = (sentences / words) 100 -
Let's first make a counte for word, sentence and letters
-
Get the average
-
Plugged it into Coleman_liau Formula
DNA profiling
Explanation of the concept
Basically the goal of DNA profiling is to have a certain piece of human body analyzed and compared by a certain person to identify if that certain piece of human body belongs to that certain person.
Matching STR counts can be used to identify who a sample of DNA belogns to
Given a sequence of DNA, how can forensic investigators identify to whom it belongs?
DNA is really just a sequence of nucleotides
Each nucleotide of DNA contains
- one of four different bases: adenine (A), cytosine (C), guanine (G), or thymine (T)
Short Tandem Repeats (STRs)
- High Genetic Diversity
- short sequence of DNA bases
- tends to repeat consecutively numerous times at specific locations
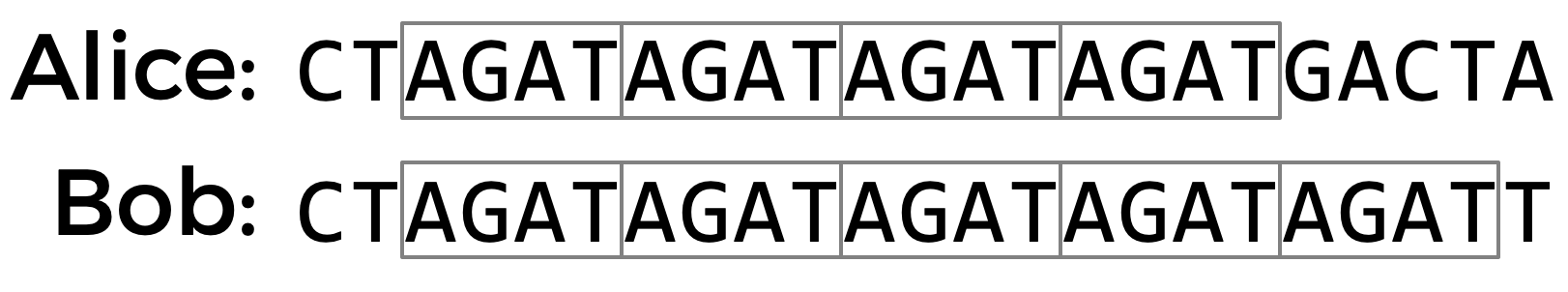
^ Alice has the STRAGATrepeated four times in her DNA,
^ Bob has the STRAGATrepeated five times.
Using multiple STRs, rather than just one,
- improves the accuracy of DNA profiling
So
IF -> two DNA samples match in the number of repeats for each of the STRs,
THEN -> the analyst can be pretty confident they came from the same person.
What does a DNA database look like?
name,AGAT,AATG,TATC
Alice,28,42,14
Bob,17,22,19
Charlie,36,18,25
.csv file above ^ // C# is the syntax to add color highlighting
Alice has a DNA sequence AGAT repeated for 28 times
Alice also has AATG repeated 42 times
Bob has TATC repeated 19 times
So given a sequence of DNA, how might you identify to whom it belongs?
Well, imagine that you have a pair of sequence of DNA
you analyzed that pair of DNA and compared it to the DNA database
-> IF you then found that
longest sequence of AGAT was 17 repeats long.
longest sequence of AATG is 22 repeats long and,
longest sequence of TATC is 19 repeats long,
-> THEN, looking at the DNA database. It matches with the DNA database of BOB. Which is a pretty good evidence that the sequence of DNA was Bob’s.
->ELSE:
it doesn’t match anyone in your DNA database,
in which case you have no match.
Your task is to write a program that will
- take a sequence of DNA
- CSV file containing STR counts for a list of individuals
- then output to whom the DNA (most likely) belongs.
What to do
Open CSV file
- opens the CSV file and read its contents into memory.
- store each line of data into a data structure
Open the DNA sequence
- open the DNA sequence and read its contents into memory.
- store each line into a data structure
Compute Longest Run
For each of the STRs
Notice that we’ve defined a helper function for you, longest_match, which will do just that!
- Where should I find out the consecutive repeats
- It says CSV file yet the file already contains how many repeats the person has
- Its most likely the
.txtfile, but I maybe wrong
- Calculate how many times each STR repeats consecutively
- Use
longest_match()function here ^ - Longest_match = DNA sequence and an STR as inputs, returns the maximum number of times that the STR repeats.
- How do we store the counts of RSA?
- How can we have it so that it remembers the data being stored and increments when needed
- And make it flexible to any length of headers
- Use
- Compare the counts against every row in the CSV file to look for a match
- What does sequence mean?
- What does subsequence mean?
- Why does this thing below matter?
--> longest run of consecutive repeats of STR in DNA sequence - Let's first understand what the existing function does
Longest Match Function
given both
inputs -> DNA sequence, and STR
Our plan is to plug in this two manually and see what happens
-
What is a DNA sequence
Ans: Like ASSAAAGGKALALAK, basically a strings of character -
What is an STR
Ans: It's the repeated value of a DNA, for instanceAAATis an str that repeats four times in DNA sequence -
Okay, so let's find out how we can read the raw DNA sequence file and assign it to a variable in python
-
We're also gonna learn how to read a .csv file into python
-
Okay so how do we find the STR?
-
I think it's on the headerrs
returns the maximum number of times that the STR repeats.
Okay so I finally understood the longest match function
dna_sequence = "AAASJFDAAASGTTTTSDAAAA"
str = "AABB"
longest_match(dna_sequence, str)
like that, but there are multiple str, so we're gonna loop it.
But each of the results is assigned to different str
Oh wait, what's gonna be the use of the results of longest_match?
Okay, so let's first understand what the "Check the database for matching profile"
- If the STR counts match exactly with any of the individuals in the CSV file, your program should print out the name of the matching individual.
- You may assume that the STR counts will not match more than one individual.
- If the STR counts do not match exactly with any of the individuals in the CSV file, your program should print
No match.
And let's plan on how to do it as well
So basically, we're gonna scan each column for each row,
Then we see if it matches with
wait wait
This is the database of .csv
name,AGAT,AATG,TATC
Alice,28,42,14
Bob,17,22,19
Charlie,36,18,25
So how exactly are we gonna compare
Hmmm, okay so let's understand what the longest_match returns
Okay, so we're gonna store the returned values of longest_match to a list.
For instance, [name, 28, 42, 14]
We're gonna remove the first index here, which is name
then we're gonna get the rows, with name_removed as well
and compare the row with the longest_match list
like
longest_match = [28, 42, 14]
database = [Alice, 28, 42, 14]
if longest_match == database[1:]:
return database[0] # returns the name
Then we just need a loop to scan each of the row
for person_dna in dna_databse:
LEZZ GOO
Final Code:
import csv
import sys
def main():
# TODO: Check for command-line usage
if len(sys.argv) != 3: # We take 2 args
print("Usage: python dna.py data.csv sequence.txt")
sys.exit()
file_csv = sys.argv[1]
file_txt = sys.argv[2]
# TODO: Read database filSTRSTRe into a variable
file_csv = open(file_csv)
file_csv = csv.reader(file_csv)
header = []STRSTR
header = next(file_csv)
rows = []
for row in file_csv:
rows.append(row)
# TODO: Read DNA sequence file into a variable
file_txt = open(file_txt)
file_txt = file_txt.read()
# TODO: Find longest match of each STR in DNA sequence
# Store every returned value to a list
list_longest_match = []
for STR in header[1:]:
STR_count = str(longest_match(file_txt, STR))
list_longest_match.append(STR_count)
# TODO: Check database for matching profiles
for person_dna in rows:
if list_longest_match == person_dna[1:]:
print(person_dna[0])
return 0
print("No Match")
return 1
def longest_match(sequence, subsequence):
"""Returns length of longest run of subsequence in sequence."""
# Initialize variables
longest_run = 0
subsequence_length = len(subsequence)
sequence_length = len(sequence)
# Check each character in sequence for most consecutive runs of subsequence
for i in range(sequence_length):
# Initialize count of consecutive runs
count = 0
# Check for a subsequence match in a "substring" (a subset of characters) within sequence
# If a match, move substring to next potential match in sequence
# Continue moving substring and checking for matches until out of consecutive matches
while True:
# Adjust substring start and end
start = i + count * subsequence_length
end = start + subsequence_length
# If there is a match in the substring
if sequence[start:end] == subsequence:
count += 1
# If there is no match in the substring
else:
break
# Update most consecutive matches found
longest_run = max(longest_run, count)
# After checking for runs at each character in seqeuence, return longest run found
return longest_run
main()
The biggest value of the repeated texts is the
-> longest run of consecutive STR
For each position
keep checking successive substrings
until STR no longe repeats
See if STR matches once, then see if STR matches twice
Okay, so what does STR contain?
What value should the STR be?
So STR is the headers of the DNA database
- Maybe what we could do is get the list of
headersin the DNA database- ^ This will act as a list of
STR
- ^ This will act as a list of
- Then starting from index 0, till the length of the STR
Printing Results
IF -> STR counts match exactly with any of the individuals in the CSV file,
THEN -> your program should print out the name of the matching individual.
ELSE IF -> STR counts do not match exactly with any of the individuals in the CSV file,
THEN -> your program should print No match.
ASSUME -> assume that the STR counts will not match more than one individual.
- Code: Implement the command line argument code
- Learn about CSV
- Learn about open and read files
Done:
Command Line Argument
Takes 1st command-line arg the name of a .csv file
- Inside of the
.csvFile -> STR counts for a list of individuals
Takes 2nd command-line arg name of.txtfile - Inside of
.txtfile -> the DNA sequence to identify
IF -> program executed with incorrect number of command-line arguments,
THEN -> program print error message of your choice (with print).
IF -> correct number of arguments are provided,
THEN -> read on ASSUME
ASSUME ->
1st argument is indeed the filename of a valid CSV file
2nd argument is the filename of a valid text file.